FDA Draft Guidance for Industry
FDA guidance for Industry which recommends the inclusion of adolescents in disease- and target-appropriate adult oncology trials to enable earlier access to investigational and approved drugs
When the histology and biologic behavior of the cancer under investigation is the same in, or the molecular target of the drug is relevant to, cancers in both adult and adolescent patients.
Provides guidance on:
- appropriate criterion for inclusion at various stages of development (first-in-human or dose-escalation, activity estimating or confirmatory)
- dosing and PK evaluations
- safety monitoring
- ethical requirements
eCRF and Standard Analyses
An eCRF is an auditable electronic record designed to capture information required by the clinical trial protocol. The design and information collected in the eCRF are then utilized by the sponsor to report the anonymized data on each trial subject either in an aggregate manner or “individual manner” in tables, listings, and figures generated via standardized analysis.
When follow-up of Growth and Development is required by the study protocol specific information may need to be collected.
We provide a list aimed to guide investigators or sponsors on specific assessments to characterize AYA growth and development in clinical trials for subsequent reporting (if deemed required by the sponsor, study protocol and/or regulatory bodies).
Tool:
Growth
- Anthropometric Measurements: height, weight, BMI (body-mass index), head circumference
- Summary display in SDS/percentile per WHO/CDC growth charts
- Bone Age from X-ray
- Disproportionate growth: sitting height/standing height ratio
- Growth Velocity
- Predicted Adult Height
Development
- Anthropometric Measurements: height, weight, BMI (body-mass index), head circumference
- Summary display in SDS/percentile per WHO/CDC growth charts
- Bone Age from X-ray
- Disproportionate growth: sitting height/standing height ratio
- Growth Velocity
- Predicted Adult Height
Others
- Anthropometric Measurements: height, weight, BMI (body-mass index), head circumference
- Summary display in SDS/percentile per WHO/CDC growth charts
- Bone Age from X-ray
- Disproportionate growth: sitting height/standing height ratio
- Growth Velocity
- Predicted Adult Height
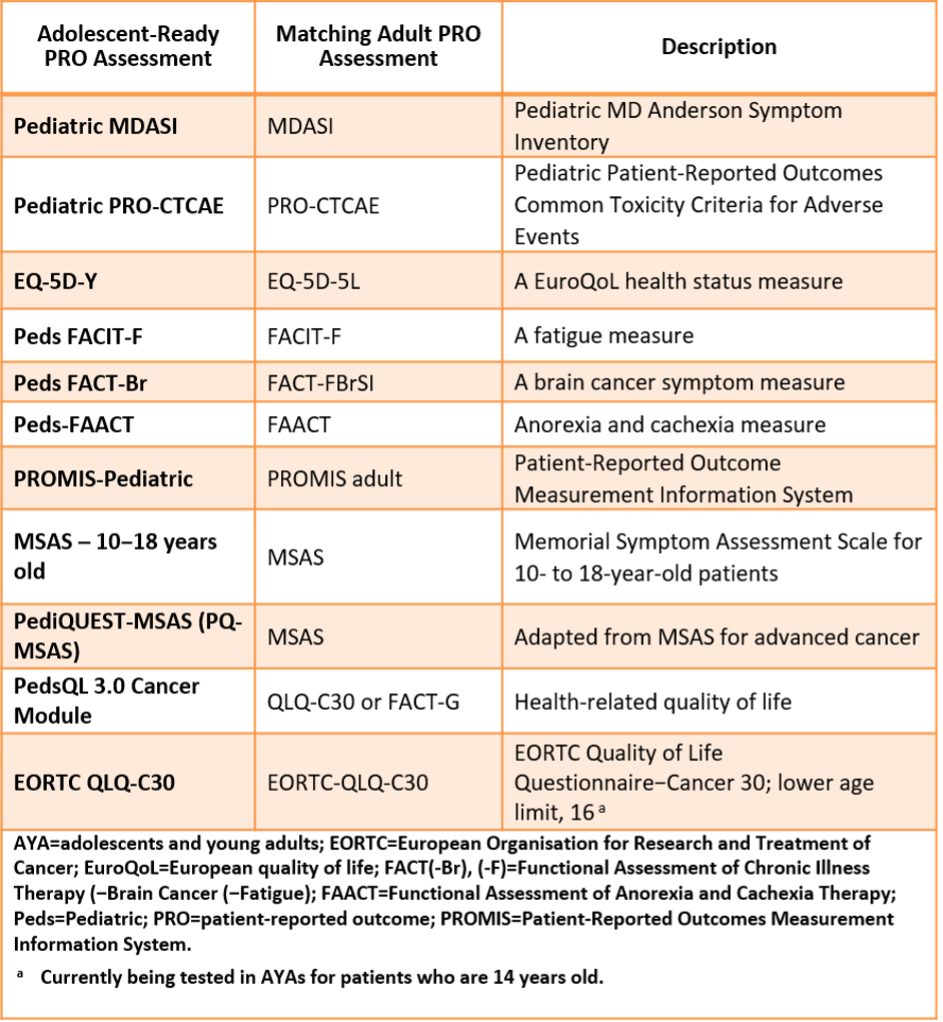
Patient Reported Outcomes (PROs)
PROs have been defined as “any report of the status of a patient’s health condition that comes directly from the patient, without interpretation of the patient’s response by a clinician or anyone else.” In other words, PRO tools measure what patients are able to do and how they feel by asking questions. These tools enable assessment of patient–reported health status for physical, mental, and social well–being.
Tool:
Example of Oncology PROs with versions suitable for AYA patients
Assent templates for adolescents
The International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) requires in module E11 that all participants are informed in a language and terms they are able to understand, and that participants of appropriate intellectual maturity should sign a separately designed written assent or ICF.
The Assent is a form of agreement of someone not able to give legal consent to participate in an activity/clinical research study. The Assent does not substitute the informed consent which is required to be signed by the parent or legal guardian and constitutes the legal documentation of understanding of the implications of taking part in the research , and agreement to take part to it.
Protocol Elements
List of protocol paragraph(s) aimed to inclusion of AYA patients:
- Assent related
Tool:
Assent related within Protocol:
“For adolescent participants unable to give their written consent, in accordance with local regulations, one or both parents, a guardian, or a legally acceptable representative must be informed of the study procedures and must document permission by signing the informed consent form approved for the study prior to clinical study participation. Each participant must be informed about the nature of the study to the extent compatible with his or her understanding. Should a participant become capable or reach the age of majority, his or her consent should be obtained as soon as possible. The explicit wish of a participant who is a minor or unable to give his or her written consent, but who is capable of forming an opinion and assessing information to refuse participation in, or to be withdrawn from, the clinical study at any time should be considered by the investigator. Minors who are judged to be of an age of reason as determined by local requirements should also give their assent. The assent should be documented based on local requirements. Continued assent should be documented when important new information becomes available that is relevant to the participant’s assent.”
“Adolescent patients age 12 years and older are allowed with signed assent and parental consent according to institutional guidelines and requirements.”
Template wording for Inclusion criteria: (Reference: J.Clin.Onc 2017; 35-3781-3787)
- Adolescent/pediatric patients age [protocol author to insert age minimum and maximum specific to the study under consideration] will be included after enrollment of adult patients once safety and toxicity have been established. Participating sites will be notified when adolescent/pediatric patient enrollment may begin.
- Adolescent/pediatric patients age [protocol author to insert age minimum and maximum specific to the study under consideration] will be included starting one dose cohort behind the current adult cohort in which there are no dose-limiting toxicities identified. Participating sites will be notified when enrollment to the adolescent/pediatric stratum may begin.
- Adolescent/pediatric patients age [protocol author to insert age minimum and maximum specific to the study under consideration] will be included in age-specific cohorts that will be staggered starting one dose cohort behind the current adult cohort in which there are no dose-limiting toxicities identified. Participating sites will be notified when each adolescent/ pediatric cohort enrollment may begin.
- Adolescent/pediatric patients age [protocol author to insert age minimum and maximum specific to the study under consideration] are included in this trial in a separate cohort that will accrue simultaneously with the adult cohort [specify age 18 and older or protocol-specific upper age limit].
Examples of HA/EC considerations on AYA
This subpage provides examples of HA requests and clarifications in relation to AYA study protocols.
This page could be divided by the type of question(s) received:
- Pre-clinical requirements
- Clinical/Protocol related
- Regulatory
- Others
